
 At the end of May I received some awful news. My former lab manager reached out with an ominous phone call: a high school student I had mentored at the University of New Hampshire had tragically passed away. His name was Evan Dube, and he was attending his first year of university at Bates College in Scotland; the students were participating in a ‘polar plunge’ and Evan collapsed on the beach after emerging from the water. He was only 19 years old.
At the end of May I received some awful news. My former lab manager reached out with an ominous phone call: a high school student I had mentored at the University of New Hampshire had tragically passed away. His name was Evan Dube, and he was attending his first year of university at Bates College in Scotland; the students were participating in a ‘polar plunge’ and Evan collapsed on the beach after emerging from the water. He was only 19 years old.
This was devastating news for me. Evan was a bright kid–the kind of student everyone hopes to find in their lab. Even though he was only in high school, I’d suggest journal articles for him to read (often talking about some pretty complex computational biology). He’d come back, having read the articles front to back, asking a slew of deeply insightful questions. I remember giving him about 5 minutes of training for our lab’s PCR protocols, and then he smoothly took over and carried out the reactions like a pro. It was like mentoring a grad student or postdoc.
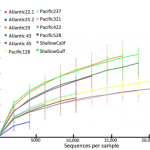
Evan was working with some of our high-throughput sequence data generated from deep sea sediments (the basis of our recent Molecular Ecology paper). As I was analyzing millions of DNA sequences and preparing our manuscript, I was interested in the overarching narrative: global patterns in biodiversity, identifying cosmopolitan species that might be present in both the Pacific and Atlantic Deep-sea. But with millions of DNA sequences you can literally ask millions of questions–each sequence has its own story.
With the broad focus of my work, things can get pretty frustrating: After processing and analyzing SO MANY sequences, our main result is basically a pie chart. Granted, a pie chart that no one has ever seen or even been able to generate before, but still a pie chart. I guess its kind of like baking up a new type of pie; pretty delicious, unless you’re really sick of eating pie.
")
So we end up slapping informative-ish names on most things. It’s a worm! This one is not a worm! This other thing is definitely an amoeba! These labels can be pretty informative…sometimes. Unfortunately there’s also always a huge (unnerving) chunk of DNA sequences that leave us shrugging our shoulders. To match DNA sequences with a species name, or even broadly lump it into a taxonomic group (and by proxy, gain an understanding of the morphology and ecological role that organism might fill in an environment), the most common approach is to compare sequence similarities between known and unknown sequences. You’re lining up two sequences, letter by letter, to see what bases match and what bases don’t. We can paste an unidentified environmental sequence into the BLAST (Basic Local Alignment Search Tool) tool at NCBI (the government-run public repository for biological sequences) and get a result that looks something like this:

Because the sequences seem to match well–the alignment is perfect–we can be pretty confident that our DNA sequence is a close relative. With this sequence, we have a 100% match to a Polychaete worm species, Aurospio dibranchiata. Unfortunately, with unknown environments we more often get results that look like this:

The percent of nucleotides that line up with the database sequence isn’t as high as we’d like (only 89%), so even though the the two sequences seem similar, there are a lot of non-matching positions in the alignment, and the BLAST scores aren’t high enough for us to automatically accept the taxonomy (for good matches, we basically copy-and-paste the taxonomy from the database sequence onto our unknown sequence). In our high-throuhput data analysis, we tend to use a 90% sequence similarity score as a minimum cutoff for accepting the taxonomy of the top-scoring BLAST result; the second sequence in particular doesn’t meet that cutoff–so, we label this as having “no match”. Before you call the Nobel Prize committee, let me just clarify: it’s not that we found some weird crazy new species in our data. This result just means that our unnamed environmental sequences doesn’t have a good match in public databases–probably because no one has generated a DNA sequence from a species that is closely related to this unidentified deep-sea creature. If we look closer, we’d find that most of these sequences seem to fit within known groups of organisms, albeit representing divergent (previously undiscovered) branches on the Tree of Life. The taxonomy from the “badly matched” sequence seems to indicate that the DNA came from a nematode, but the statistical score is too low for us to be confident about this assignment.
In another scenario, it’s quite common to have uninformative taxonomic assignments tacked on to your query sequences, because researchers who deposit data into public sequence databases like NCBI don’t always put useful labels on their DNA. Someone, somewhere might have sequenced the SAME species–they found a DNA sequence that looks exactly like your sequence, with the BLAST sequence similarity of 100%–but if the researcher didn’t label the sequence with any useful taxonomy (like “nematode” or “paramecium”), then we can’t link an informative name with the DNA. This situation is frustratingly ubiquitous–there are far too many datasets where everything is labelled as “unclassified environmental sequence”. Researchers who went into an environment and sequenced a bunch of DNA, but weren’t interested in (or didn’t have the capacity for) attaching taxonomic names to the sequences they generated. An example from our dataset, where the label of “uncultured eukaryote” is absolutely useless:

So all of this brings me back to Evan. For his summer lab project, we wanted to delve deeper into the world of enigmatic deep-sea sequences that were labelled as “no match” or “unclassified environmental sequence”. Since high-throughput sequencing technologies only return short DNA barcodes (100 to 400 bases long), we thought that perhaps we could generate gene-length sequences (~1000 bases long) that would be more informative for studying molecular evolution, and inferring the relationship between these divergent sequences and known taxa in public databases. We could take this approach because our baseline dataset was generated LIKE A BOSS. In our deep-sea study, we simultaneously amplified two different DNA barcodes from our environmental pool, located at either end of the 18S rRNA gene:

Evan was pulling out sequences from both ends of the gene labelled as “no match” or “environmental sequence”, and trying to match up sequences exhibiting similar overall proportions (=relative abundances) at both loci. He’d look a bit deeper into the BLAST results (pouring through a list of ~50 close relatives in the database, instead of just looking at the top-scoring hit) and postulate the Phylum each sequence belonged to. We then paired up “matching” sequences from either end of the gene, hoping that similar taxonomy and abundances meant we had generated two DNA barcodes from a single species. The next step was to design sets of PCR primers that were highly specific to these environmental sequences; using customized primers designed to bind either end of the gene, we wanted to amplify the 18S rRNA gene from *one* species, starting from the complex genomic mixture of deep-sea environmental DNA. This was a crazy idea. We had no idea if it would work. We had no idea if we were even matching up sequences from the same species.
Evan got reeeeeeealllllyyyy good at doing PCR. Since we were taking a shot in the dark, we wanted to exhaust all our options before giving up completely. Every forward primer had to be tested with every reverse primer (and vice versa) to make sure we hadn’t mis-matched sequences from the “same” species. And of course designing PCR primers is notoriously difficult anyways, so even if we were matching up things correctly our awesome idea might be complete rubbish anyway, for chemical and logistical reasons. Evan became a PCR reaction mixologist and a connoisseur of agarose gels.
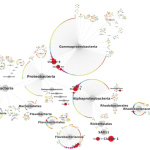
The moral of this story: sometimes the ambitious project you give your undergrad or high school student is miraculously successful. We got positive results. Full-length 18S rRNA sequences. AND we could use these to build a badass tree. So now, drumroll please…here’s some unpublished data for you guys. The blue sequences in the figure below are our reference sequences, pulled down from public databases. The red sequences are the full-length 18S rRNA genes Evan skillfully managed to amplify from our messy pool of deep-sea DNA.

So what does this mean, biologically? Well for one, an extremely talented student got some serious training in cutting-edge research. Secondly, it seems we were able to isolate DNA from a really cool group of species from deep-sea mud, identifying a divergent clade that has never before been sequenced.
Once we placed our gene sequences onto the Tree of Life, we could see that our red sequences (labelled “Contig”) fell within a group of eukaryotes known as the Labyrinthulids. This is a supercool group of protists known as slime nets (people used to think these things were fungi)–the main cell body glides along the sticky, filamentous strings as it uses these extensions to slurp up nutrients.

During this project, Evan learned the importance of tree-based (phylogenetic) approaches, and the pitfalls of taxonomic assignments that rely on BLAST-based sequence similarity alone. As the field of high-throughput sequencing progresses, the outlook is slowly getting better for putting names on millions of environmental DNA sequences. We’re now moving towards (more robust) phylogenetic approaches to assign taxonomy to unknown sequences, where short sequences reads are placed onto a reference guide tree constructed out of full-length gene sequences. Short reads are “tested” and scored at every position in the tree, and inserted into the tree at the best-scoring position. Little by little, tree-based methods such as these are slowly reducing our reliance on BLAST comparisons and their inherent limitations.

Hearing the news about Evan has made me think a lot about science. The beach where Evan took his final swim was probably littered with some of the very enigmatic taxa that he had been seeking in our datasets. This post is a tribute to Evan: his research in New Hampshire, his memory, and his unrealized potential. I’m so grateful for the opportunity to work with such a talented young man, and extremely saddened by his loss. My thoughts go out to his family, friends, and fellow students. Rest in peace, Evan.



What a loss that this bright lad died so young.
Dr. Bik – Thank you so much for this amazing tribute to our Evan! He was such a wonderful young man in every way possible he lived his life to the fullest. It is a tribute to his life that even though it was a few years ago that he worked with you, that he left such an impact on you. Thank you for putting the results of the research out there too. It is another way that his life will continue to leave a mark.
with deepest gratitude,
Eileen Dube (Evan’s Mom)