
This past week I was visiting the University of Delaware to attend the 3rd Skate Genome Annotation workshop, sponsored by the IDeA Network for Biomedical Research Excellence (INBRE) Program from the National Center of Research Resources at the National Institutes of Health.
As the title suggests, we’re looking at real data from the genome project of the Little Skate (Leucoraja erinacea). Why is this cool? Well firstly because all marine animals are totally awesome (Even vertebrates…I guess). Secondly, the Little Skate is often used as a model organism for understanding the human biology. L. erinacea is a Chondrichthyan fish, a primitive jawed vertebrate that branched off early from all living vertebrate species. The Little Skate functions like any typical vertebrate, possessing an adaptive immune system and a pressurized circulatory system (plus it grows fairly easily in tank); this species has significantly enhanced our knowledge of human physiology, immunology, stem cell and cancer biology, pharmacology, toxicology, and neurobiology. Having a complete genome sequence for the Little Skate will have huge benefits for developmental biology and biomedical research, and will also help to decipher evolution in sharks, rays, and higher vertebrates . With 49 chromosomes and an estimated genome size of 3.42 billion base pairs (slightly larger than the human genome), this species has considerably less genetic matter than other cartilaginous fishes (the genome of the dogfish shark, another closely related model organism, is double the size).
”]
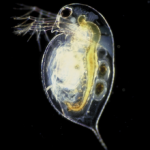
The most awesome thing about the Little Skate is its phenomenal power of regeneration. As in, it can grow back organs and amputated limbs. One of the big reasons for sequencing the genome is to characterize the genetic pathways and patterns of gene expression that enable this wound-healing response. If the Little Skate is an ancient vertebrate, then you could reason that limb regeneration is an ancestral trait that was subsequently lost in higher-level vertebrates. If we understand which genes turn on, could we eventually learn how to switch on limb regeneration in humans?
”]![Wound healing response in the Little Skate [Photo Credit: Ben King at Mount Desert Island Biological Lab]](https://www.deepseanews.com/wp-content/uploads/2011/05/Screen-shot-2011-05-28-at-3.16.58-PM.png "Screen shot 2011-05-28 at 3.16.58 PM")
A smaller genome is also cheaper to sequence (making scientists happy) and equals a better chance of a successful assembly. Although we hear about new genomes being sequenced on a weekly basis, this stuff is hard work. Take the human genome—our Homo Sapiens genome assembly is pretty damn good (nevermind ten years old already), yet there are STILL bits and pieces of sequence that don’t seem to fit in anywhere. Imagine you’re thisclose to completing a 2,000 piece puzzle, but you’ve got a bunch of holes and your extra pieces are all the wrong shape and size (e.g. you probably wrongly jammed in a piece somewhere in there…now you have to find it and swap it out). In a genome, we’ve got genes flanked by repetitive ‘junk’ DNA. A lot of times we don’t have long enough sequence bridges to span the gap of these repetitive regions; with the newest technologies each sequenced strand of DNA (what us biologists refer to as ‘reads’) is 100-150 base pairs long, but repetitive regions can be thousands of bases in length. The Skate Genome project currently has sequenced over 3 BILLION reads and given us 59x coverage of the genome (meaning every position in the genome has theoretically been sequenced 59 times) but there STILL isn’t enough data for a good assembly. The current assembly has the genome spit into 3 million contigs (longer stretches of DNA stuck together), with the longest contigs around 21,000 bases in length. Curse you, repetitive elements! Nevertheless, we’re making slow and steady progress; the data that we do have is helping to train undergrads, postdocs, and new genomicists in genome assembly and protein identification. Our undergrad from UNH was dreaming about gene annotation by the end of the week…



Does a lizard’s tail have anything related to this type of regeneration? Besides my question though, I think deep sea life is easily the most interesting type of life. I’d like to see more organisms that are clear, and you can see the organs inside of them. I can’t tell from that first picture if this is clear or not. Either way, that’s badass. :)
I find it interesting that the little skate has such a short genetic code compared to spiny dogfish, since current taxonomy suggests that skates and rays came from the dogfish line of sharks. That has to be taken with a grain of salt because our understanding of the taxonomy of rays is always changing. Also, I actually never knew that skates could regenerate parts of their wings.
Hi Hunter, Yes – any type of limb regeneration is considered a ‘wound healing response’. Humans can repair wounds but not whole limbs, so we have a sort-of imperfect regeneration ability. The big question is figuring out if limb regeneration is something that the ancestor of all vertebrates had (meaning Humans lost the ability to do this somewhere along the line), or if this response has evolved lots of times in different groups of animals. Anyway, I found a cool podcast that talks more about tissue regeneration, check it out if you want to know more: http://www.sciencenetlinks.com/sci_update.php?DocID=419
P.S. Skates aren’t see-through, but lots of jellyfish are (my favourite deep-sea animal!)
There are various new and innovative television shows that offers the crazy entertainment.
If it’s entertainment you seek, why not check out the hotels spa for a day of total rest.
Technology, entertainment, and the impact of watching live cricket matches
is an important milestone that will go a long way in
encouraging, as well as, promoting the live streaming of live cricket games.