
![]()
If you’ve ever talked to me in person for more than 5 minutes, I’ve probably mentioned the !#$%*ING AWESOMENESS of high-throughput sequencing. Frankly, I’m a bit obsessed. If my life were an SAT analogy, it would be Dr Bik:Sequencing platforms as Teenage Girls:Twilight. My gorgeous Illumina never sleeps (runs 2 weeks straight for a 125 bp paired end run); you must feed it with precious fluid (but damn those reagents are expensive). And most importantly, it sparkles (pyrosequencing = flashes of light as individual nucleotides are incorporated).

But I’m not the only one who’s infatuated. Sequencing’s evolution has ushered in a veritable revolution for aquatic research. Its not that us marine biologists were being lazy all these years–beer is an integral part of the research process of course–but we just didn’t have the tools needed to characterize the vast undiscovered biodiversity residing in the oceans. Driven by fundamental advances in DNA sequencing technology, it is now possible to use an approach called metagenomics :

The basic definition of metagenomics is the analysis of genomic DNA from a whole community; this separates it from genomics, which is the analysis of genomic DNA from an individual organism or cell…definitions have varied to include any study whereby a whole community is analyzed, e.g., directed studies of 16S rDNA diversity from an environment to isolation and analysis of total DNA from environmental samples without prior cultivation (Chen & Pachter 2005). [Gilbert & Dupont, 2011]
Some pretty cool stuff is coming out of recent studies. Thanks to deep-sequencing, tracking natural selection through population genomics is becoming the status quo. It looks like regional environments can shape the rather pliable genome contents of common water column bacteria such as Pelagibacter ubique, and there is now evidence for geographic isolation even in microbial species. We also get metabolic insight: for example, Prochlorococcus bacteria living in iron-poor waters of the tropical Pacific possess amazingly efficient, streamlined cellular pathways to extract this important trace element from their surroundings.
Furthermore, all those nice textbook graphics that illustrate nutrient cycles are probably waaaaaay too simplistic: Microbe X produces this metabolite which is taken up by Species Y, etc etc. We thought we had a pretty good understanding of Phosphorus cycling. Except that we didn’t. Metagenomics now tells us that “biologically inert” phosphonates are actually some of the main molecules utilized by oceanic microbes. The classical view had been developed using microbes easily cultured in the lab—microbes which, in real ecosystems, represent only a minute fraction of the community.

What’s remarkable is that all this groundbreaking knowledge has come from literally a drop of seawater. Every study churning out millions of DNA sequences is analyzing less than 1% of the genetic material in a sample. Since 2004 when next-generation sequencing platforms such as 454, Illumina and SOLiD really took off, scientists have amassed ~400 billion base pairs of microbial DNA—so, in 7 years we’ve looked at only 3% of the nucleotides present in 1 milliliter of seawater. High-throughput sequencing platforms are like the Hubble telescope for biologists—really exciting and powerful, but once you turn them on and look through the lens at all those new galaxies to catalogue, your first response is “Oh, damn…”
I forgot to mention that metagenomic studies have pretty much ignored eukaryotes up to this point. (That’s where my lab comes in–we’re fighting for the underdogs!):
One of the underlying reasons why scientists using metagenomic tools have forsaken the eukaryotes is the cost required compared with that for prokaryote-focused projects. Even the smallest free-living eukaryote, the photosynthetic prasinophyte Ostreococcus, has a genome five times larger than that of an average marine bacterium (Derelle et al. 2006, Palenik et al. 2007). At the more extreme end of the spectrum, some dinoflagellate genomes appear to be much larger than the human genome (Hackett et al. 2005). Confounding this issue, eukaryotic genomes are far less gene-dense than those of bacteria and archaea, meaning that equivalent sequencing efforts will yield much more information for prokaryotes. Another hurdle is the combination of the inherent diversity of the eukaryotic superkingdom, the lack of reference genomes, and the phylogenetic complexity of eukaryotic genomes. [Gilbert & Dupont, 2011]
In other words, everyone knows the eukaryote researchers (= me) are pretty screwed at this point. Microbial biologists jumped on the DNA bandwagon back in the 1970s, and now they have decades of genetic information and resources at their fingertips. On the other hand, some guy just found out that nematodes actually have DNA (well, not really, but you get the idea). Taxonomy has been, and will continue to be, a powerful force for phyla that can be visualized under a microscope. Unfortunately, this reigning morphological tradition can also hinder progress–Deep sequencing is powerful, but it also needs powerful tools; you can’t annotate a million sequences by hand. Eukaryote researchers have a long wish list of software and scripts that need developing, but we’re making vast strides every day. We’re bioinformatic ninjas.

Reference:
Gilbert, J., & Dupont, C. (2011). Microbial Metagenomics: Beyond the Genome Annual Review of Marine Science, 3 (1), 347-371 DOI: 10.1146/annurev-marine-120709-142811



You have to like this article, great read: http://www.plosone.org/article/info%3Adoi%2F10.1371%2Fjournal.pone.0018169
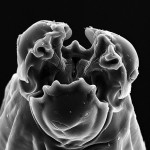
everything ends up in the deep sea, and yes, nematodes are the most diverse metazoan in the deep (Ant)arctic according to this study!
Oooh, I didn’t know you were a sequencing nerd too ;) Although, one point of contention, the Solexa machine uses reversible dye termination to read the bases. I don’t know if you do the sequencing runs yourself, but the cleavage mix at position 6 cuts off the dye after every cycle to regenerate the 3′ end for the next round of base incorporation. 454 is a pyrosequencer :)
*drool*
Thanks for the insightful blog about the article. We really want to help improve the Eukaryota analysis capable with metagenomic research – the microbial eukaryotes especially are an immensely valuable part of the ecosystem and no model would be complete without understanding their role in shaping functional potential and metabolite turnover – especially across seasonal cycles.
Keep up the cool blogging. @gilbertjacka